一个重磅炸弹的中国接力
与驻沪记者小康合作的一篇文章,Frank刚把文章修回来,增加了许多专业和关键的说法。特别提及中国的临床前试验数据还未完全被国际认可(如OECD成员国),这意味着用于全球注册目的临床前的研究将可能在国外进行,值得关注。
一个重磅炸弹的中国接力
一方面是跨国药企寻求研发外部合作的战略践行,一方面是科研院所对于基础成果转化为临床药物的渴求,两者的碰撞或将催生中国的“重磅炸弹”。
6000万美金,有望成为重磅炸弹的一项蛋白抗肿瘤药物专利,这是赛诺菲-安万特日前与上海生命科学院所开展的一项合作交易。通过签订专利与技术许可合同,赛诺菲-安万特公司已获得上海生科院的授权,将在全球范围内进一步开发该项抗肿瘤新药物。
赛诺菲与上海生科院的合作在某种意义上,有如一场研发接力,充分体现了药物研发的全球化分工合作。在一个“重磅炸弹”可能形成路径的背后,除了凸显企业与科研院所的联盟意义,对于中国而言,随着全球药物同步开发进程的加快,需重新审视在创新链条上的角色定位,而后期临床出开发能力以及整体创新环境迫待加强与改善。
最好的时间点
“这是对我们实行合作式研发战略的最好鼓励。” 赛诺菲-安万特全球研发副总裁、亚太研发总裁江宁军难掩兴奋。两年前,赛诺菲-安万特与上海生科院签订战略合作伙伴协议,锁定在糖尿病、癌症和中枢神经系统疾病领域开展合作研究。而此次专利许可就源于双方在肿瘤领域的研究进展,是一项由上海生科院生物化学与细胞生物学研究所发明的蛋白抗肿瘤药物专利技术,作用机理在于阻断肿瘤的血管生成,有望治疗多种癌症,其中包括肝癌等在中国高发的癌种。
“做基础研究是个长期的过程。我们生科院对这个项目已经做了多年的基础研究工作,已有一定基础。与企业的合作,加速了开发进程。”上海生科院副院长吴家睿指出。
据了解,该药物是针对新药耙的首创新药,是癌症治疗领域的突破,该项原始创新成果的专利保护范围将涉及几十个国家和地区。这意味着如果研究获得最终成功,该成果可望成为癌症治疗领域的新的重磅炸弹。
良好的前景吸引赛诺菲-安万特下注。根据合同约定,上海生科院生化与细胞所发明的蛋白抗肿瘤药物的专利与技术授权赛诺菲-安万特公司实施, 6000万美元并不是一次性付清,而是先付一笔入门费,然后按照每一阶段的进展支付,当所有临床指标和注册完成后6000万美元全部支付完毕,之后生科院还将每年获得一定百分比的销售额提成。
事实上,这正是体现效益共享与风险共担的国际惯例。原上海生科院副院长,上海医药临床研究中心主任甘荣兴评价说:“根据化合物的重要性,创新药的价值会随着研究上市进程的进度而梯级上升。如抗肿瘤药,一个新化学体完成临床前的价值大约在500万美元,临床I期完成的价值大约在1500~2 000万美元。最后卖给国际大型制药公司的价格有可能高达2500~5000万美元。因此,这种合作模式对于中国科研单位而言,必须运用知识产权战略,辅助那些资金来源困难的研究项目做一些早期临床研究使项目增值,既能卖得好价钱又不会卖得太早。”在他看来,此次上海生科院对项目的转让正处于一个最合适的阶段,而且在没有临床前和临床研究按数据的情况下能卖到6000万美元价格很合理。该药前期发现全部在上海生命科学院进行,专利转让后由赛诺菲-安万特进行全球开发。
“我们正在为临床试验做准备工作,包括对该生物制剂的制造和质量控制的开发以及临床前安全评价等。”据江宁军透露,从临床前到进入I期临床研究大致需要两年的时间。在进展顺利的前提下,药物的最后上市会在十年以内。
后期开发待突破
“我们科研机构擅长的是前期基础研究,现在的一个重大课题就是转化,即怎样把基础研究和应用结合起来,尽快的把基础研究成果应用到临床,这就需要与企业进行合作。而对企业而言,要实现在短时间内最有效的研发,同科研单位合作是个非常好的方式。未来大型企业与生命科学研究院所的联盟是一个必然发展方向未来”吴家睿指出。事实上,上海生科院目前已有一个20多人专业从事知识产权转让的团队,该团队人员构成都有生物医药或法律背景,全程跟踪科学家的药物发现,专门进行项目评估、申请专利及洽谈项目。
据了解,上海生科院对此项目的运作完全按照美国产学研的模式在进行,该交易也被业界视为非常传统和经典,“科学发明不等于有专利,有专利不等于能许可。首先科学发现要全球认可;第二是要拿到全球知识产权;第三是卖一个好价钱。这是一个很长的过程。”甘荣兴说。
正如江宁军所指出的:“中国科研院所前期药物筛选能力很强,而跨国公司则有后端的药物开发优势,有强大的临床研究团队,这是一个很好的合作模式。”甘荣兴相信“这种交易会越来越普遍,体现了药物创新的分工合作。”在他看来,这种交易的买家主要将集中在跨国企业,原因在于“跟整个研发水平有关,这种药物后期全球开发需要国际化运作,临床研究也是以欧美国家药品上市注册标准进行,而国企整体水平、资金、人才尤其是政策环境都远远没达到,只有跨国巨头才会有兴趣和实力将如此早期阶段的化合物收入囊中。”
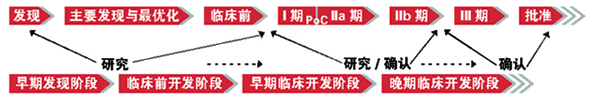
对江宁军而言,目前最迫切的是推进临床前研究的GLP进程和国际有关组织的认可。由于中国的临床前试验数据还未完全被国际认可(如OECD成员国),这意味着用于全球注册目的临床前的研究将可能在国外进行,“中国早期药物发现能力、化学合成能力非常强,但是从前期到临床研究前这一阶段,与国际还存在差距。”
据透露,目前赛诺菲正同步在中国、其他亚太国家及其公司内部进行安全评价性实验,尽快推进首次人体试验是目前的重点。
江宁军认为,从临床研究的阶段来看,跨国公司不会出于成本的考虑将首次人体试验阶段放在中国操作,因为其投入与Ⅱ、Ⅲ期大规模的试验相比是非常低的,而且可供选择的国家和地区很多。
但如果中国能够更早地参与一个新药的前期研发,将更早地介入到后面的Ⅱ、Ⅲ期试验,因为整个新药开发是连贯性的。
“如果中国仅仅是参与Ⅲ期,只是入组病人,实施试验,那么中国就成了一个巨大的CRO,整个试验的核心没有带到中国,在知识层面和理念上并没有得到很大提升。在这一层面上,我们希望中国能参与全部的开发环节,这样真正意义上的新药可以完全从中国产生。”江宁军说。
据介绍,不久前升级为亚太研发中心的赛诺菲上海研发中心将调动其亚太临床资源,完全以欧美药品注册标准进行后期开发。
“中国需要在创新链条上定位角色,加强后期开发能力。如何在发达国家完成临床试验,实现数据认可和销售分成,对于那些具有独家创新品种的研发企业和科研院所来说,我们的路还有很长。”甘荣兴指出。
{kind=link}